Biography
Juan Luis Mateo Cerdán is Associate Professor in the Department of Computer Science at the University of Oviedo. His research is focused on Machine Learning and Data Mining with application to biological problems, specially in Genomics.
Before he was at the University of Heidelberg from 2010 to 2016. There, in the Centre for Organismal Studies, was part of Joachim Wittbrodt’s lab where he worked along with biologists applying his data analysis skills to different problems. At the same time he acquired an invaluable knowledge and experience in several kinds of molecular biology experiments.
His research and teaching activity began at the University of Castilla-La Mancha, in the Sistemas Inteligentes y Minería de Datos (SIMD) group led by José Antonio Gámez Marín and José Miguel Puerta Callejón.
- Bioinformatics
- Genomics
- Data Mining
- Machine Learning
-
PhD in Advanced Computer Technology, 2010
University of Castilla-La Mancha
-
Master in Advanced Computer Technology, 2008
University of Castilla-La Mancha
-
Engineering in Computer Science, 2002
University of Castilla-La Mancha
-
Technical Engineering in Computer Science, 2000
University of Castilla-La Mancha
Featured Publications
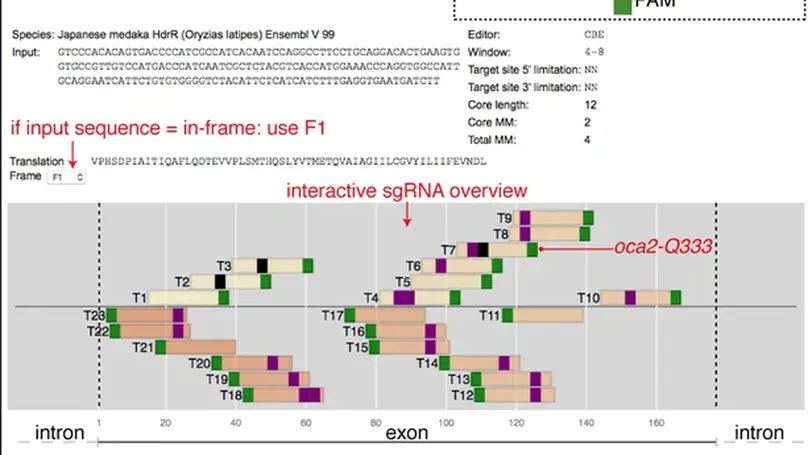
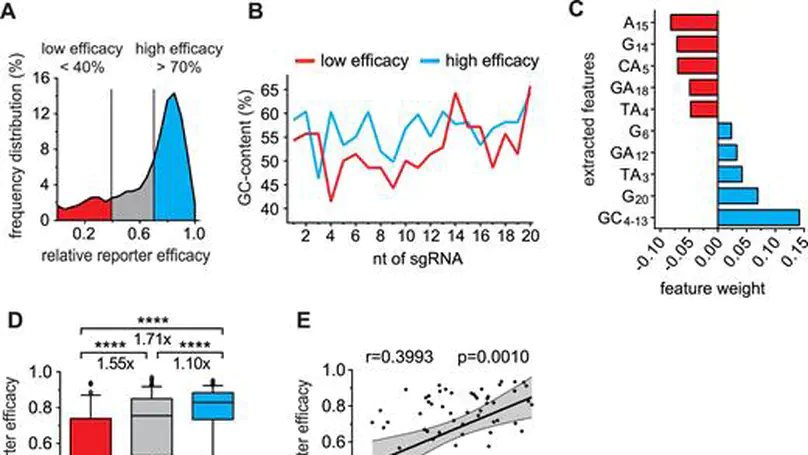
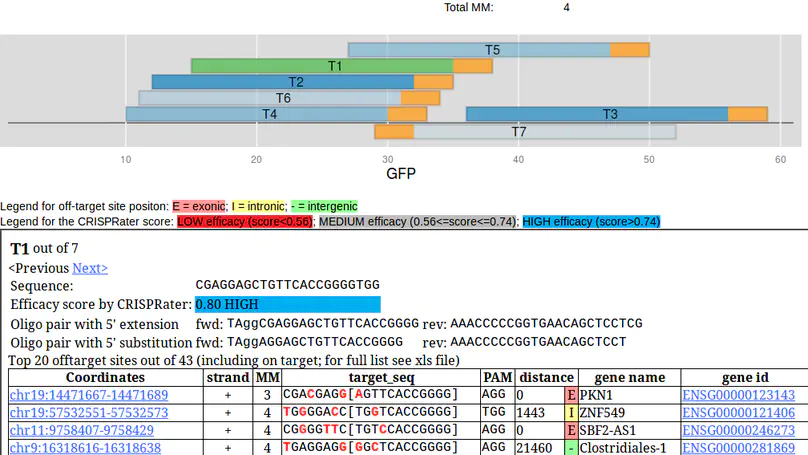
Engineering of the CRISPR/Cas9 system has opened a plethora of new opportunities for site-directed mutagenesis and targeted genome modification. Fundamental to this is a stretch of twenty nucleotides at the 5’ end of a guide RNA that provides specificity to the bound Cas9 endonuclease. Since a sequence of twenty nucleotides can occur multiple times in a given genome and some mismatches seem to be accepted by the CRISPR/Cas9 complex, an efficient and reliable in silico selection and evaluation of the targeting site is key prerequisite for the experimental success. Here we present the CRISPR/Cas9 target online predictor (CCTop, http://crispr.cos.uni-heidelberg.de) to overcome limitations of already available tools. CCTop provides an intuitive user interface with reasonable default parameters that can easily be tuned by the user. From a given query sequence, CCTop identifies and ranks all candidate sgRNA target sites according to their off-target quality and displays full documentation. CCTop was experimentally validated for gene inactivation, non-homologous end-joining as well as homology directed repair. Thus, CCTop provides the bench biologist with a tool for the rapid and efficient identification of high quality target sites.
Recent Publications
Software
ACEofBASEs (a careful evaluation of BaseEdits) standalone
ACEofBASEs is a tool to determine sites to be editted with the CRISPR/Cas9 technology in a input sequence and predict its potential off-target sites. The online version of ACEofBASEs is available at http://aceofbases.cos.uni-heidelberg.de/
The standalone version has a command line interface designed to allow search of large volume of sequences and higher flexibility. Also advisable if the target genome is not publicly available.
Access ACEofBASEs standalone from my bitbucket site.
CCTop (CRISPR/Cas9 target online predictor) standalone
CCTop is a tool to determine suitable CRISPR target sites in a given query sequence(s) and predict its potential off-target sites. The online version of CCTop is available at http://cctop.cos.uni-heidelberg.de/
The standalone version has a command line interface designed to allow search of large volume of sequences and higher flexibility. Also advisable if the target genome is not publicly available.
Access CCTop standalone from my bitbucket site.
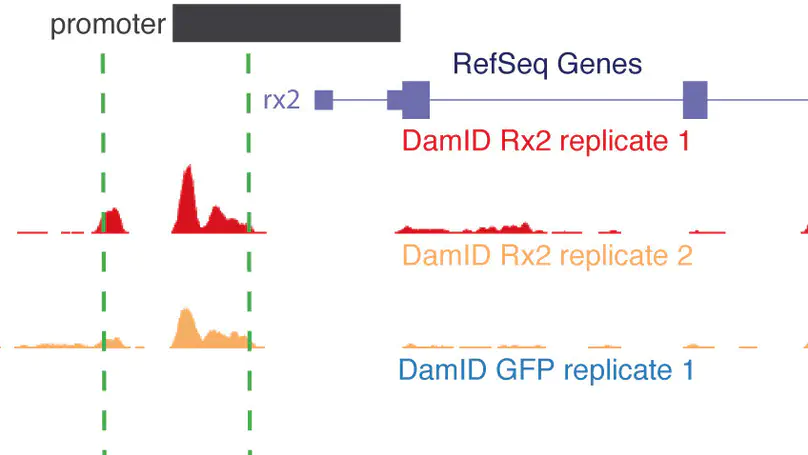
iDEAR (iDamID Enrichment Analysis with R)
iDEAR identifies the binding profile of a transcription factor (or any DNA binding protein) from iDamIDseq data according to our publication in Development.
Access iDEAR from my bitbucket site.
Teaching
Currently I teach at the University of Oviedo the following subjects:
- Sistemas Inteligentes (Grado en Ingeniería Informática del Software)
- Programming Methodology (Grado en Ingeniería Informática en Tecnologías de la Información)
- Bioinformática (Máster Universitario en Biotecnología Aplicada a la Conservación y Gestión Sostenible de Recursos Vegetales)
Previously
- Repositorios de Información (Grado en Ingeniería Informática del Software)
- Sistemas Operativos (Grado en Ingeniería Informática en Tecnologías de la Información)
- Tecnologías y Paradigmas de la Programación (Grado en Ingeniería Informática en Tecnologías de la Información)
- Arquitectura del Software (Grado en Ingeniería Informática del Software)
At the University of Heidelberg I’ve taught these subjects (2014-2016):
- Introduction to Statistical Analysis for Biologists with R (Bachelor in the Faculty of Biosciences)
- Data Analysis with R (Bachelor in the Faculty of Biosciences)
- Introduction to Genetics (Bachelor in the Faculty of Biosciences)
- Statistics for Biology: design, analysis and visualization (Core course in the HBIGS International PhD Program in Molecular and Cellular Biology)
At the University of Castilla-La Mancha I taught these subjects (2006-2010):
- Minería de Datos (Ingeniería Informática)
- Bioinformática (Ingeniería Informática)
- Metodología de la Programación (Ingeniería Informática)
- Estructuras de Datos y de la Información (Ingeniería Informática)
Contact
- +34 985 10 2943
- Jesús Arias de Velasco s/n, 33005 Oviedo, Spain
- Geology Faculty, 7th floor, Room 7-1